
The CE marking process is a pivotal step for manufacturers looking to distribute their:
- medical devices,
- machinery,
- other related products within the European Economic Area (EEA) and
- other regions recognising CE standards.
This mark signifies compliance with the essential requirements of the relevant European health, safety, and environmental protection legislation.
Entering into this process reveals the complexity and necessity of understanding specific requirements, documentation, and the strategic approach to obtaining CE marking.
Understanding the intricacies of this process, including the specific requirements, documentation, and flowcharts, is essential for manufacturers aiming to navigate the complexities of compliance successfully. For further assistance in navigating these requirements, consider visiting Qreg, which offers detailed guidance and support for achieving CE marking.
Table of Contents
Overview of the CE Marking Process Flowchart

The journey to achieving CE marking begins with a structured process flowchart, which serves as a comprehensive guide for manufacturers.
This flowchart delineates the initial step of identifying applicable directives or regulations, such as:
- The Medical Devices Directive (MDD), the In Vitro Diagnostic Devices Directive (IVDD), and their successor regulations,
- The Medical Devices Regulation (MDR) and the In Vitro Diagnostic Devices Regulation (IVDR), respectively.
Navigating through these regulations is critical for ensuring Regulatory Compliance, which encompasses not only adherence to specific standards but also a broader commitment to meeting all legal requirements for market entry.
The CE Marking Process
Identify the Applicable Directive:
The first step involves identifying which EU Directive or Regulation applies to your medical device.
- The most relevant are the Medical Devices Directive (93/42/EEC),
- The In Vitro Diagnostic Devices Directive (98/79/EC),
- The Active Implantable Medical Devices Directive (90/385/EEC),
- Which are being replaced by the Medical Devices Regulation (MDR 2017/745) and the In Vitro Diagnostic Medical Devices Regulation (IVDR 2017/746).
Classify Your Device:
Medical devices are classified according to the level of risk they pose to patients and users.
The classification ranges from Class I (lowest risk) to Class III (highest risk), with the classification determining the level of assessment required.
Choose a Conformity Assessment Route:
Depending on the device’s classification, manufacturers must choose an appropriate conformity assessment route, which may require the involvement of a Notified Body, an organization designated by an EU country to assess the conformity of certain products before being placed on the market.
Compile a Technical File or Design Dossier:
Manufacturers must compile a technical file or design dossier demonstrating that the medical device complies with the relevant requirements.
This includes details on design, manufacture, and performance of the medical device, along with the results of risk analysis and clinical evaluations.
Implement a Quality Management System (QMS):
For most classes of devices, implementing a QMS compliant with ISO 13485 is required. This system ensures ongoing compliance and product quality.
Assessment by a Notified Body (if required):
For all but Class I devices (unless sterile or with a measuring function), an assessment by a Notified Body is required. This may involve an audit of the QMS and a review of the technical documentation.
Declaration of Conformity:
This legal document, drafted by the manufacturer, declares that the device meets all EU regulatory requirements. By signing this document, the manufacturer takes full responsibility for the product’s compliance.
Affix the CE Mark:
Once all steps have been successfully completed, the CE Mark can be affixed to the medical device or its packaging, along with the identification number of the Notified Body involved in the assessment process.
Post-Market Surveillance and Vigilance
After a device has been placed on the market, manufacturers must implement post-market surveillance to ensure continued compliance and to monitor the safety and performance of the device.
Any incidents or safety concerns must be reported to the relevant authorities under the EU vigilance system.
Adapting to Post-Brexit Changes: CE Marking and UKCA Marking
Adapting to the changes brought about by Brexit, particularly concerning CE marking and UKCA marking, involves understanding the new regulations and ensuring compliance.
CE Marking (Conformité Européenne):
- CE marking indicates that a product complies with EU regulations and can be freely traded within the European Economic Area (EEA).
- Post-Brexit, CE marking continues to be recognized in the EU, but the UK has introduced its own marking system, UKCA, for goods placed on the market in Great Britain (England, Wales, and Scotland).
UKCA Marking (UK Conformity Assessed):
- UKCA marking is required for goods placed on the market in Great Britain.
- It covers most goods that previously required CE marking.
- Northern Ireland continues to recognize CE marking, and UKCA marking is not required there, except for certain products.
- UKCA marking involves conformity assessment by a UK-approved body, similar to CE marking.
To adapt to these changes:
Understand Applicability:
- Determine whether your products require CE marking, UKCA marking, or both.
- Consider geographical markets and regulatory requirements.
Update Documentation and Labelling:
- Update technical documentation, labels, and packaging to reflect the correct marking for the intended market.
- Ensure that products bear the appropriate marking and accompanying documentation.
Compliance Assessment:
- Ensure that your products meet the applicable UK or EU standards and regulations.
- Engage with notified bodies or conformity assessment bodies as needed.
Transition Period:
- Keep abreast of transition periods and deadlines for compliance.
- Be aware of any changes or updates to regulations during the transition.
Stay Informed:
- Regularly monitor updates from relevant authorities, industry associations, and legal experts to stay informed about any changes or developments in regulations.
Seek Expert Advice:
- Consider consulting with regulatory experts or legal counsel specializing in product compliance to ensure thorough understanding and compliance with the new requirements.
Adapting to post-Brexit changes may require initial investment in updating processes and documentation, but ensuring compliance with CE and UKCA marking regulations is crucial for continued market access and successful trade in the UK and EU markets.
The UK’s departure from the EU introduced the UK Conformity Assessed (UKCA) marking for products placed on the market in Great Britain. Despite this change, the CE marking continues to hold relevance for products sold in Northern Ireland and the EEA.
Manufacturers must now navigate the dual pathways of CE and UKCA marking for products intended for both markets, adapting to the distinct requirements of each. In the midst of these adjustments, the “MDR transition” represents a critical pivot point for medical device manufacturers, aligning with new regulatory frameworks to maintain market compliance.
Mastering the CE Marking Process
Achieving CE marking is more than a regulatory complexity; it’s a testament to a product’s safety, quality, and compliance with the highest European standards.
Manufacturers must engage in continuous education and adaptation to regulatory changes, including the nuances of specific device directives and the evolving landscape post-Brexit.
- By meticulously following the CE marking process flowchart,
- Maintaining comprehensive technical documentation, and
- Adhering to conformity assessment protocols,
- Manufacturers can successfully navigate the complexities of CE marking,
- Ensuring their products are poised for success in the European market and beyond.
This enhanced understanding and strategic approach to the CE marking process not only facilitate compliance but also underscore a manufacturer’s commitment to delivering products that meet the highest standards of safety and performance, ultimately benefiting consumers and healthcare providers alike.
Navigating Changes: The Transition to MDR and IVDR
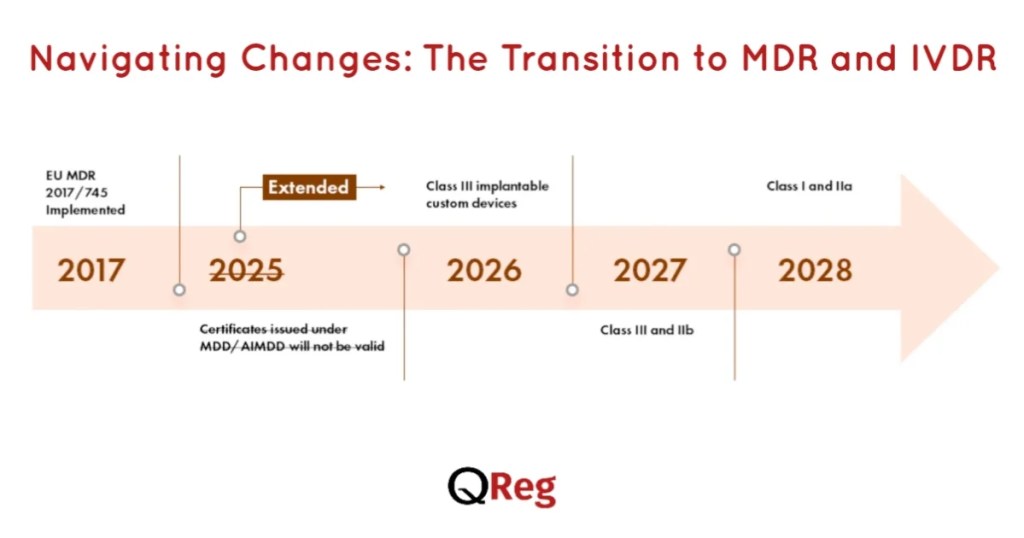
Manufacturers must be aware of the transition from Directives to Regulations (MDR and IVDR), which introduce more stringent requirements and greater scrutiny of medical devices.
This includes enhanced clinical evaluation, post-market surveillance, and transparency measures.
FAQs (Frequently asked questions)
What is CE Marking?
CE marking, short for “Conformité Européenne,” is a certification mark that indicates conformity with health, safety, and environmental protection standards for products sold within the European Economic Area (EEA).
It is a mandatory requirement for certain products to be legally placed on the market in the European Union (EU) and European Free Trade Association (EFTA) countries.
Which Medical Devices Need CE Marking?
In the context of medical devices, CE marking is mandatory for most devices intended for medical use within the EEA.
This includes:
- a wide range of products such as diagnostic equipment,
- surgical instruments,
- implants, and
- other medical devices.
All medical devices intended for sale in the EEA need CE marking. Devices are classified according to the potential risks associated with their use, from low risk
(Class I) to high risk (Class III).
Do I Need a Notified Body for CE Marking?
Not all devices require a Notified Body.
Class I devices (with some exceptions) can be self-certified. Classes IIa, IIb, and III generally require the involvement of a Notified Body for the conformity assessment procedure.
For certain classes of medical devices, involvement of a notified body is mandatory.
Notified bodies are organisations designated by EU member states to assess the conformity of certain products with EU regulations.
They conduct conformity assessments, which may include testing, auditing, and examining technical documentation, to ensure compliance with relevant EU directives or regulations.
How Long Does the CE Marking Process Take?
The timeline can vary significantly depending on the device class, the completeness of the documentation, and the need for clinical investigations.
It can range from a few months for simpler devices to several years for more complex devices requiring clinical studies. Notified bodies are organisations designated by EU member states to assess the conformity of certain products with EU regulations.
They conduct conformity assessments, which may include testing, auditing, and examining technical documentation, to ensure compliance with relevant EU directives or regulations.
How Much Does CE Marking Cost?
- Costs can vary widely based on device class,
- The complexity of the device,
- The extent of required clinical evaluations,
- The cost of obtaining CE marking for medical devices varies depending on factors such as the classification of the device,
- The involvement of a notified body, and
- The services required to demonstrate compliance.
Costs may include testing fees, assessment fees charged by notified bodies, consultant fees (if applicable), and expenses related to documentation preparation.
Is CE Marking Valid Indefinitely?
No,
CE marking is not valid indefinitely. It requires:
- Ongoing compliance with the relevant legislation,
- Including post-market surveillance,
- Periodic safety update reports for higher-class devices, and
- Re-assessment if significant changes are made to the device or its intended use.
What Happens if a Device Fails to Comply After Receiving CE Marking?
If a device is found not to comply with the relevant regulations after receiving CE marking, it may be subject to recall, penalties, or other enforcement actions.
Manufacturers are required to ensure ongoing compliance through vigilant post-market surveillance and reporting.
To ensure compliance with CE marking requirements for medical devices, manufacturers should carefully follow the applicable regulations, engage with notified bodies when necessary, maintain comprehensive documentation, and monitor changes in regulatory requirements.



